
This is really a continuation of my post in which I tried to lay out the basic chemical reactions associated with the uptake of 

Figure showing the relative concentrations of the different inorganic carbob compounds plotted against pH.
I’ll start by showing the relative concentration of the different inorganic carbon compounds plotted against pH. I should point out that in the previous post I suggested that the equations are solved by setting the total Dissolved Inorganic Carbon (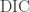
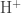
![{\rm [H^+]}](https://s0.wp.com/latex.php?latex=%7B%5Crm+%5BH%5E%2B%5D%7D&bg=ffffff&fg=333333&s=0&c=20201002)
![pH = -log {\rm [H^+]}](https://s0.wp.com/latex.php?latex=pH+%3D+-log+%7B%5Crm+%5BH%5E%2B%5D%7D&bg=ffffff&fg=333333&s=0&c=20201002)
When the pH is small, the

Figure showing how the atmospheric CO2 concentration varies with temperature for fixed DIC and TA.
Something else you can do is look at how the atmospheric 

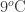

Figure showing how the DIC (top), Revelle factor (middle) and pH (bottom) vary with atmospheric CO2 (pCO2).
The last thing I was going to discuss was how the 

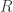
![R = \dfrac{\frac{\Delta p{\rm CO_2}}{p{\rm CO_2}}}{\frac{\Delta DIC}{DIC}} = \dfrac{DIC}{DIC - {\rm \frac{([HCO_3^-] + 2[CO_3^{2-}])^2}{([HCO_3^-] + 4[CO_3^{2-}])}}}.](https://s0.wp.com/latex.php?latex=R+%3D+%5Cdfrac%7B%5Cfrac%7B%5CDelta+p%7B%5Crm+CO_2%7D%7D%7Bp%7B%5Crm+CO_2%7D%7D%7D%7B%5Cfrac%7B%5CDelta+DIC%7D%7BDIC%7D%7D+%3D+%5Cdfrac%7BDIC%7D%7BDIC+-+%7B%5Crm+%5Cfrac%7B%28%5BHCO_3%5E-%5D+%2B+2%5BCO_3%5E%7B2-%7D%5D%29%5E2%7D%7B%28%5BHCO_3%5E-%5D+%2B+4%5BCO_3%5E%7B2-%7D%5D%29%7D%7D%7D.&bg=ffffff&fg=333333&s=0&c=20201002)
The red line is the above calculation, while the blue line is the Revelle factor determined from the data plotted in the upper panel. They don’t exactly match, but they’re pretty close and have the same form. If you would like the python script that did the calculation and produced the figures, it is here.
The middle panel in the figure on the right shows that, for current conditions, the Revelle factor is about 10. This means that the fractional change in atmospheric
This has got a little long, but hopefully illustrates some of the carbonate chemistry of seawater. There are a number of aspects that I haven’t considered, such as the timescale over which atmospheric 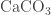
Dissolution into ocean water sequesters 70–80% of the CO2 release on a time scale of several hundred years. Chemical neutralization of CO2 by reaction with CaCO3 on the sea floor accounts for another 9–15% decrease in the atmospheric concentration on a time scale of 5.5–6.8 kyr. Reaction with CaCO3 on land accounts for another 3–8%, with a time scale of 8.2 kyr. The final equilibrium with CaCO3 leaves 7.5–8% of the CO2 release remaining in the atmosphere. The carbonate chemistry of the oceans in contact with CaCO3 will act to buffer atmospheric CO2 at this higher concentration until the entire fossil fuel CO2 release is consumed by weathering of basic igneous rocks on a time scale of 200 kyr.
As I’ve already said, I’m no chemist, so hopefully I’ve got this about right. There is probably much more that could be said, and some subtleties that I may have missed. If anyone would like to elaborate, or clarify, feel free to do so via the comments. This is really mostly a chance for me to learn something while, hopefully, not confusing others 🙂 .
Update:
Eli has a post about The Revelle factor and also includes some discussion about the role of calcium carbonate.
Eli has another post about the biological pump.

This is very informative. Thanks.
That was the intent 🙂
ATTP … Archer et al, in the paper you reference include an important consequence/ observation, following Fig. 4/ Eqn. (3):
“In order the terrestrial the terrestrial biosphere or silicate rock weathering, for example, to restore atmospheric CO2 to its preanthropogenic value, we require uptake not only of the atmospheric CO2 excess, but the entire fossil fuel CO2 load, including that which has reacted with CaCO3, reducing the cumulative net atmospheric release to zero …”
Richard,
Yes, and I think that isn’t appreciated. It’s not sufficient to simply remove what is in the atmosphere; to return to pre-industrial concentrations would require removing all of our emissions, whether that removal is natural (silicate rock weathering) or is done by us.
great content, always a pleasure to read your stuff.
the largest issue in my mind is the effect of stratification, a slowing of the AMOC will prevent more waters rising from the deep column that was last exposed to the atmosphere during pre-industrial times. My estimate is that an 85% slowdown of this current would lead to a 7% decline in total annual CO2 sequestration by the biosphere. This is, in my mind, one of the major potential understated impacts of a further warming world. We are already seeing this effect happening and if our atmospheric fraction drops from 54% to 35% we will receive an additional 2 billion metric tonnes of CO2 accumulation in the earth’s atmosphere per year (above today’s values).
Jai,
Interesting comment. I hadn’t considered that, but certainly seems that it could play a big role in the ocean uptake of CO2.
Although it seems blindingly obvious as soon as you briefly think about it, this (essentially Richard’s point above) was one of the more sobering, frustrating things I realized when I read this Hansen et al contribution in 2013. And I like the way it is communicated:
Under equilibrium conditions a negative CO2 pulse, i.e., artificial extraction and storage of some CO2 amount, decays at about the same rate as a positive pulse (Fig. 4A). Thus if it is decided in the future that CO2 must be extracted from the air and removed from the carbon cycle (e.g., by storing it underground or in carbonate bricks), the impact on atmospheric CO2 amount will diminish in time. This occurs because carbon is exchanged among the surface carbon reservoirs as they move toward an equilibrium distribution, and thus, e.g., CO2 out-gassing by the ocean can offset some of the artificial drawdown. The CO2 extraction required to reach a given target atmospheric CO2 level therefore depends on the prior emission history and target timeframe, but the amount that must be extracted substantially exceeds the net reduction of the atmospheric CO2 level that will be achieved. We clarify this matter below by means of specific scenarios for capture of CO2 (From the section “Carbon Cycle and Atmospheric CO2”)
Figure 4A (highlights mine) makes the point that removing a slug of CO2 from the atmosphere yields the mirror image of the decay scenario for adding CO2 to the atmosphere.:
url of image: http://imagizer.imageshack.us/a/img921/6785/ASm9ix.png
You can only go so far with a chemical analysis of the problem because biological activity plays a huge role.
Eli,
Indeed, that was what I was implying at the end of the post, but I’m only just starting to get to grips with the chemistry; biology is still beyond me 🙂
@rust – I think it’s cheaper to extract CO2 from the ocean. In which case you wouldn’t have the problem you describe above. Extracting CO2 from the ocean also helps deal with ocean acidification.
-1,
I think the point is that you still have to ultimately remove as much as has been emitted, whether you take it directly from the atmosphere, or from the ocean.
Thanks for your posts. I note that your calculation of the effect of temperature upon atmospheric CO2 is consistent with mine: you have 9C rise causing 280 ppmv to 400 ppmv, I have 12C causing a 40% increase, and the ice-core CO2-temperature measurements show 12C causing 190 ppmv to 300 ppmv.
In your first post, you appear to be assuming that Henry’s Law applies only to the CO2(aq) species, but I could be mis-reading you. To clarify, for pure water and CO2 we have:
pCO2 = k * ([CO2(aq)] + [H2CO3] + [HCO3-] + [CO3–])
where k is a constant and pCO2 is the partial pressure of CO2 gas above the water, and I’m using square brackets for concentration of each species. If we dissolve CaCO2 in the water, we have have:
pCO2 = k * ([CO2(aq)] + [H2CO3] + [HCO3-] + [CO3–] – [Ca++])
I would call the concentration on the right side of each equation the “dissolved CO2 concentration”, but you may be using that phrase in a different way, to refer only to the H2CO3 concentration, in the graph at the top of this post. I can see why you would interchange the two names: I have read a dozen chemistry chapters on this subject in the past week, and they interchange the terms H2CO3*, H2CO3, CO2(aq), Dissolved CO2, and Total Carbon Concentration. I think are lax about these terms because CO2(aq) is the dominant species in systems of pure water and CO2.
The Revelle Factor appears to express the fact that most of the carbon in the ocean is not from atmospheric CO2, but from dissolved CaCO3 and other sources. You have the Revelle Factor around 10 for the ocean today. If the total carbon mass in the top layer of the ocean is 2000 GT, then only 200 GT of that carbon is dissolved CO2 taking part in the CO2 cycle. Is that right?
I note that dissolved CaCO3 does not affect the rate at which CO2 molecules strike the ocean surface and are dissolved (an exothermic reaction 19 kJ/mol), nor does it change the rate at which CO2 emerges from the ocean by thermal excitation (endothermic 19 kJ/mol). The concentration of CO2 in the atmosphere will be in proportion to the mass of dissolved CO2 in the ocean. If we double the CO2 pressure in the atmosphere, the rate at which CO2 dissolves will double. Thus I see no means by which the CaCO3, nor any change in pH, can affect the linearity of the carbon cycle’s diffusion equations. Do you agree? If not, can you show me some equations that express the non-linearity generated by the ocean chemistry?
Kevan,
As I understand it, it does only apply to aqueous CO2.
No, I think the Revelle factor only applies to fractional changes. In other words, if the atmospheric CO2 concentration changes by some fraction, by what fraction does the total Dissolved Inorganic Carbon change. To actually do some kind of calculation, you then do need to know the actual amount of inorganic carbon in both reservoirs. My understanding, is that its the upper ocean that is relevant, but I haven’t found a good source for the depth of the upper ocean. The entire ocean contains about 36000 Gt of Dissolved Inorganic Carbon, so if you assumed half (say 20000 GtC) that would probably be reasonable.
No, I think you’re ignoring that it’s buffered. As I said above, Henry’s Law applies only to the aqueous CO2, not to the total inorganic CO2.
That’s this post. You can download one of the python scripts and play around with it.
“As I understand it, it does only apply to aqueous CO2.”
By your assumption, only aqueous CO2 has any significant probability of making the leap into the atmosphere. The liberation of aqueous CO2 takes 19 kJ/mol, or 0.02 eV per molecule. The liberation of some other species takes 0.02 eV per molecule plus the energy, E, required to transform the species back into aqueous CO2 before it makes the leap into gaseous form. By your assumption, E is so large that other species cannot transform back into CO2 and jump into the atmosphere. But if that’s the case, there is will be no equilibrium between the species: there will be no aqueous CO2, because whenever an aqueous CO2 molecule turns into another species, it will never go back. And yet we observe that there most certainly is an equilibrium between the various species, so we have arrived at a contradiction. Your assumption cannot be true.
Henry’s law applies to the total dissolved carbon dioxide concentration.
The rest of your post makes sense to me now. If we assume Henry’s Law applies only to one of the several species of dissolved carbon, then we conclude, incorrectly, that when the equilibrium shifts away from that species, the Henry’s Law constant for CO2 is being altered significantly, and we would conclude that the diffusion of CO2 into the ocean is non-linear, and our CO2 sticks around for 100 years. But that’s not the case. Neither the pH of the ocean, nor changes in the equilibrium between the dissolved carbon species, nor the addition of CaCO3, can effect the statistical mechanics of CO2 entering the ocean (vastly exothermic) and leaving the ocean (vastly endothermic, 0.02 eV compared to thermal energy of 0.025 eV).
Kevan,
These aren’t my assumptions; this is the standard chemical reactions for the dissolution of CO2 in seawater. It includes all the different reaction rates and we expect it to tend to a state of equilibrium (i.e., the forward and backward reactions balance). It’s really not just me.
Except it’s not incorrect.
Actually, we expect some fraction of our emissions to stick around for thousands – ultimately it’s expected to take maybe 200000 years to return to pre-industrial levels if we leave it to nature.
Here’s what happened last time – 200ka to get rid of the mess:
Source: Zachos et al. (2008) An early Cenozoic perspective on greenhouse warming and carbon cycle dynamics
Correction: CO2 emerging is not “vastly endothermic”, it’s “mildly endothermic”.
It is impossible for Henry’s Law to apply only to one species of dissolved CO2. I believe I demonstrated this using a simple thermodynamic argument. The fact that other people believe that Henry’s Law applies only to one species of dissolved CO2 is their personal choice. Their belief in no way alters the reality.
In the chemistry texts that I have been reading, they apply Henry’s Law to the entire dissolved concentration. See here, for example:
Click to access Section5.pdf
I can see where the confusion comes from: the terminology in the above chapter is [H2CO3*] referring to total dissolved concentration, but they drop the star sometimes, and they have this delightful line: “where H2CO3 = H2CO3 + CO2 (aq) = H2CO3*”.
Kevan wrote “In your first post, you appear to be assuming that Henry’s Law applies only to the CO2(aq) species, but I could be mis-reading you. “
AFAICS this is the mainstream scientific position. A standard textbook on this kind of thing (nearly 1300 Google citations, is:
Rcihard E. Zeebe and Dieter Wolf-Gladrow, “CO2 in Seawater: Equilibrium, Kinetics and Isotopes”, Elsevier Oceanography Series, volume 65, 2001. ISBN 0-444-50946-1
Regarding Henry’s law it states (page 67, eqn 1.5.68):
[CO_02] = K_0(T,S) \cdot PCO_2
where PCO_2 is the partial pressure of CO2 in the atmosphere K_0(T,S) is a temperature and salinity dependent constant and [CO_2] is “The CO_2 concentration in surface waters”. Now this would be deeply misleading if Henry’s law actually depended on DIC, if that was the case, they would have written DIC or [DIC]. However, equation 1.1.9 (page 4) relates [CO2] and DIC:
[CO_2] = DIC/(1+K^*_1/[H+] + K^*_1K^*_2/[H+]^2)
Which makes it very clear that Zeebe and Wolf-Gladrow were not using [CO2] to mean DIC in eqn 1.5.68 (the K’s are the constants that define the equilibrium distribution of carbon-containing species).
“Your assumption cannot be true.”
We discussed this on your blog. If you persist in thinking that all apparent contradictions are caused by someone else’s mistake, rather than entertain the possibility that the misunderstanding may be yours then the rate at which you can learn will be very slow as you are willfully blinding yourself to half of the available area where the mistake may lie. Now the fact that the Revelle effect is very well understood and accepted by the research community doesn’t mean that it is right and you are wrong, but it does mean that common sense suggests it is hubris to assume Revelle and all who have followed him are wrong, rather than you, without taking the time to read (and possibly reproduce their results/models). Before you mention “argument from authority” (yet again), this isn’t one, I am just recommending caution and a bit of self-skepticism as being a better way to learn and to resolve disagreements.
By coincidence, Richard Zeebe is a co-author of Zachos et al. (2008) linked above.
Kevan wrote “It is impossible for Henry’s Law to apply only to one species of dissolved CO2. I believe I demonstrated this using a simple thermodynamic argument. The fact that other people believe that Henry’s Law applies only to one species of dissolved CO2 is their personal choice. Their belief in no way alters the reality.”
I’m sorry but the hubris/irony of this statement is quite astonishing.
“By your assumption, only aqueous CO2 has any significant probability of making the leap into the atmosphere. The liberation of aqueous CO2 takes 19 kJ/mol, or 0.02 eV per molecule. The liberation of some other species takes 0.02 eV per molecule plus the energy, E, required to transform the species back into aqueous CO2 before it makes the leap into gaseous form. By your assumption, E is so large that other species cannot transform back into CO2 and jump into the atmosphere. But if that’s the case, there is will be no equilibrium between the species: there will be no aqueous CO2, because whenever an aqueous CO2 molecule turns into another species, it will never go back. And yet we observe that there most certainly is an equilibrium between the various species, so we have arrived at a contradiction. Your assumption cannot be true.
There are several unsupported assertions here, and there is no attempt to do any actual calculations (or even looking up of the numbers). I am no chemist, but ionic solutions form because the overall energy of the dissociated ions is lower than that of water and the ionically bonded molecule, so it doesn’t seem that surprising if the required energy is fairly high. Also the if the dissolution depends partially on the [CO2] in the solution, then that is what is stopping it coming out of solution directly already. I don’t see the argument that this would mean there could be no equilibrium between species, that seems to be based on a rather naive view of the kinetics etc. (my understanding is also naive, but I don’t have the hubris to think that is all that is necessary). Now I don’t know if Kevan is right or wrong, but his argument falls way below the level required for me to be tempted to accept it, especially if it means that the world experts have totally misunderstood the basics of the oceanic carbon cycle since the 1950s.
Now I don’t particularly want Kevan to explain his argument in more detail, as the problem is that he is not taking time to understand the mainstream position first, and hence is bound to run into “things I don’t know I don’t know” territory. Understanding Zeebe and others is likely to be a much more productive approach.
Kevan quoted (http://lawr.ucdavis.edu/classes/ssc102/Section5.pdf) thusly: “where H2CO3 = H2CO3 + CO2 (aq) = H2CO3*”
Note that [H2CO3] + [CO2(aq)] is not DIC. Zeebe and Wolf-Glashow have (eqn 1.1.1)
[CO2] = [CO2(aq)] + [H2CO3]
However [H2CO3] is “true carbonic acid” (i.e. a molecule) and this is not the same thing as dissociated [H+] and [HCO3-] ions, with the equilibrium given by eqn 1.1.3:
CO2(aq) + H2O H2CO3 HCO3- + H+ CO3– + 2H+
DIC is [CO2] + [HCO3-] + [CO3–]
There is very little true carbonic acid, relative to CO2(aq) (about 0.3% according to Zeebe and Wolf-Glashow), which is why it seems to be frequently neglected (only learned about this wrinkle this morning). Thus it seems that Kevan may not have understood what he was reading, but the quote from his book/paper in no way suggests that Henry’s law depends on “the entire dissolved concentration”!
Sadly Kevan’s lack of self-skepticism brings him to the wrong conclusion:
“I can see where the confusion comes from: the terminology in the above chapter is [H2CO3*] referring to total dissolved concentration, but they drop the star sometimes, and they have this delightful line: “where H2CO3 = H2CO3 + CO2 (aq) = H2CO3*”.”
so it appears that the confusion turned out to be Kevan’s AFAICS.
Sorry, the in the second equation seem to have been interpreted as unknown HTML tags 😦
If you consider CO2 and O2 you get this plot:


O2 data for Alert Alaska. CO2 Barrow
and
Co2 and O2 Scales are different but it does show that CO2 and O2 is linked. I.e. it is not simply being absorbed into the ocean (which would not affect O2 levels.
Antarctic and arctic CO2 plots show a 6 month phase shift – Arctic having a much greater yearly change.
Looking at the timings of peaks and troughs it is not the obvious culprits causing them.
Decay releasing CO2 sequestered during growing season is not going to happen in arctic autumn where the increase in co2 is max and the temperatures are lower.
I would propose that it is perhaps phytoplankton respiration/photosynthesis! – dark co2 out o2 in light co2 in o2 out.
This plot shows that there is a peak phytoplankton growth in summer at the poles which is significantly higher than spring or autumn growths.
Phytoplankton in sunlight photosynthesises using CO2 and creating O2. In the dark O2 is used in phytoplankton respiration and CO2 is released.
Decomposition requires warm temperatures for the bacteria to work. At the end of summer temperatures are falling rapidly so decomposition will not release CO2 rapidly and certainly would not continue into December. This suggests that spring growth and autumn decomposition would not cause the CO2 / O2 humps.
Click to access 10AR.pdf
Experiment: I think the second equation should have been:
CO2(aq) + H2O ⇄ H2CO3 ⇄ HCO3- + H+ ⇄ CO32– + 2H+
Kevan,
Dikran’s already said most of it, but you seem to be ignoring that all the different reactions (for dissolving CO2 at least) are included above. One (Henry’s Law) relates the exchange between the ocean and the atmosphere which involves atmospheric CO2 and dissolved CO2 (mainly aqueous CO2 but with a small amount of H2CO3- – which is often ignored) and then the reaction of CO2 with water to form bicarbonate and the reaction of bicarbonate to form carbonic acid (although Nick Stokes suggests that the latter two should be seen as one reaction). These are then solved together to determine the steady state concentrations. You can’t simply assert that this is wrong. You need to actually illustrate this somehow. It might be possible to write it all as one reaction with a single coefficient, but that would probably then have some kind of temperature dependence and maybe also a dependence on DIC.
Kevan, Henry’s law only applies to [CO2], period. You can test this using a simple solution of 1 M sodium carbonate in a closed container with a nitrogen atmosphere – make it such that the pressure inside remains constant. Now measure the P(CO2) above this solution. Subsequently acidify this solution using some strong acid to pH 4 or 5, and redo the measurement. Your P(CO2) will increase, despite the fact that DIC is the same for the two solutions.
In a similar experiment, take seawater and add a little bit of calciumchloride. Measure the P(CO2) before and after. Watch it increase, despite the fact that calciumcarbonate has precipitated out of solution. That is, your DIC decreased, because of carbonate precipitation, but your P(CO2) increased. You can also measure pH and see it go down.
Now, unless I completely misunderstood what you are trying to say, neither observation fits with your claim that Henry’s law applies to DIC, and not [CO2] alone.
Yes, Kevan is wrong, as ATTP, dikran and Marco have been explaining. A few late comments.
Henry’s Law, as so often, is misused. It describes the equilibrium partition of a soluble gas between two phases. But the situation of interest here is quite out of equilibrium. There is a one-way flux of CO₂ downward. Equilibrium can only be considered when there is eventually a balancing upward flux. It’s true that there is an apparently constant airborne fraction, which sounds like partitioning. But that constancy is a result of the particular dynamics of exponential rise, not equilibrium.
You can say that in a smaller scale (top surface layer, maybe mm) there is equilibrium, because transport there is relatively fast. Then Henry’s Law kind of applies, and it governs the ratio to dissolved CO₂, not DIC. Chlorine has a Henry’s Law coefficient, but you can’t count the chloride in the sea when applying it (fortunately for mariners). Now it’s true that you can’t easily distinguish between CO₂ and H₂CO₃, and they are often both counted. That works because there is an equilibrium relation between the two
CO₂+H₂O⇌H₂CO₃
with law of Mass Action
[CO₂]*[H₂O]/[H₂CO₃]=K
Since [H₂O] is constant (and usually omitted) that means that [CO₂] and[H₂CO₃] are proportional, and you can define a Henry’s Law coef with one or both. But when CO2 reacts with carbonate:
CO₂+CO₃⁻⁻+H₂O⇌2HCO₃⁻
the mass action expression is not linear, because the reagent carbonate is consumed.
Marco,
Thank you for describing those experiments. I spent many happy hours as a boy blowing corks out of test tubes with those reactions. In the sodium carbonate experiment, the CO2 is generated by the reaction between Na2CO3 and acid, with the CO2 coming from the dissolved Na2CO3, so it’s not exactly true to say that the “dissolved inorganic carbon” remains the same, but I think you may have meant “hardly changed”, and I agree with all else that you have said.
You did misunderstand me, but I think that’s because I have not been clear enough with my terminology. Let us define the [Dissolved CO2] as the concentration of all carbon species in the water that would be absent if the partial pressure of CO2 above the water were zero. Thus, in your sodium carbonate example:
[Dissolved CO2] = [CO2(aq)] + [H2CO3] + [HCO3-] + [CO3–] – [Na+]/2
I claim that Henry’s Law applies to this [Dissolved CO2] concentration, by an argument of statistical mechanics. But my claim is not important, for the following reason.
As Dikram mentions, and according to what I have read, the [Dissolved CO2] is 99.84% CO2 (aq). The remaining 0.16% is carbonate ions. The first figure in ATTP’s post above, which is the same as Figure 5.1 below, shows the relative abundance of the three main species of carbonate in water for various pHs. The left-most graph is labeled “Dissolved CO2”, but that is incorrect. It should be labeled “H2CO3”.
Click to access Section5.pdf
I can see how “Dissolved CO2” and “H2CO3” get confused: it’s the terminology used by chemists. They write [H2CO3*] to mean “CO2 (aq) plus all carbonate species”. When the left-most graph is labelled “Dissolved CO2”, we get the impression that the equilibrium between CO2 and the water will be profoundly affected by pH. When we label it correctly as H2CO3, we see that it’s only the relative abundance of the H2CO3 in 0.16% of the dissolved CO2 that is being affected. At a fixed pH and temperature, Henry’s Law applies to 98.84% of the dissolved CO2 concentration, even if we do not accept that it applies to the 0.16% of carbonate ions.
Having said that, the carbonate ions do affect the pH, and if the pH drops, I’m guessing that some of the CaCO3 will release CO2 into the atmosphere, in the same way vinegar causes Na2CO3 to release CO2. I have not figured out how to calculate the magnitude of that effect. Maybe it’s in the post above and I just have to look more closely.
Kevan,
Maybe read Nick’s comment.
No, look at the figure in my post. The dashed line is our current pH. It’s mostly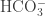 (86%). It’s only about 0.5% aqueous
(86%). It’s only about 0.5% aqueous  .
.
ATTP, Well, I’m certainly confused. Is that black line the H2CO3 carbonate concentration or is it the CO2 (aq) concentration, or are they the same thing? What fraction of CO2 dissolved in water is CO2 (aq)? Yours, Kevan
Kevan,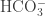 , and about 13% is
, and about 13% is  .
.
The black line in my figure is CO2(aq). As Dikran’s quote indicates, it should probably be CO2(aq) + H2CO3, but H2CO3 is only about 0.3% of the Dissolved CO2, so it doesn’t make any real difference. At a pH of 8.1, about 0.5% of the DIC is CO2(aq), about 85.5% is
Ah, very good: that makes sense of the terminology. Thank you. So, as the partial pressure of CO2 increases, the ocean acidifies, and the proportion of the dissolved inorganic carbon that is CO2 (aq) increases. (I guess you are using the DIC to distinguish from organic carbon chemicals that don’t play a part in the ionic equilibrium.) If only CO2 (aq) can make the jump to gaseous CO2, then the concentration of CO2 (aq) is set by the partial pressure of CO2 in the atmosphere. It may be that other carbonates can transform into CO2 (aq) and back again, but the rate at which CO2 is emitted is a function of both the energy required AND the number of CO2 (aq) molecules available, so even though we can compose a Henry’s Law constant to account for the movement of these carbonates into CO2 (aq) and then into the atmosphere, this constant changes with pH. If instead we ignore the movement of carbonates into gaseous CO2, and compose a constant only for CO2 (aq), the constant remains the same, which is much more convenient. I’ll read your posts again now that you have enlightened me, and get more out of them. Yours, Kevan
“As Dikram mentions, and according to what I have read, the [Dissolved CO2] is 99.84% CO2 (aq). The remaining 0.16% is carbonate ions.”
Just to be clear, I didn’t say that the remaining is carbonate ions, but H2CO3 (carbonic acid) which is a molecule, not the dissociated ions.
It would help if Kevan paid a bit more attention to what is written as I could hardly have made this point any more clearly:
“However [H2CO3] is “true carbonic acid” (i.e. a molecule) and this is not the same thing as dissociated [H+] and [HCO3-] ions,”
I can strongly recommend the Zeebe and Wolf-Gladrow book, as it seems to have been written with non-specialists in mind, so it explains quite a lot of stuff that isn’t in the papers. One thing I found out that I hadn’t previously known was that H+ ions are almost absent in seawater, because it forms hydrate complexes with H2O to form things like H3O+ and H904+, which seems to make sense as water is a polar molecule. Perhaps this is why H+ and HCO3- ions don’t readily combine to form H2C03 (which can subsequently spontaneously dissociate to form CO(aq) which can readily leave solution), because to do so the HCO3- would need to strip the H+ ion from the hydrate first. Perhaps that is the flaw in the simple energetics argument, i.e. it isn’t just about energetics, but kinetics? I’m not a physical chemist, so I don’t know, but that seems a reasonable (partial) explanation. It appears that salinity also has an effect because of interactions between the carbonate ions and the ions from the dissolved salts.
When we are dealing with the real world (and especially the bomb-carbon issue) it is also important to remember that we don’t live on a waterworld (although sadly we do have Kevin Costner), and I suspect one of the reasons that the decay time of a pulse of 14CO2 is rather longer than the residence time is because the seasonal cycling of carbon from primary production/respiration is mostly releasing the 14CO2 that it took in this year within only a year or two, which is too fast compared to the half-life of 14C for it to affect atmospheric concentrations of 14CO2, but it certainly does have a big effect on the residence time.
” If only CO2 (aq) can make the jump to gaseous CO2, then the concentration of CO2 (aq) is set by the partial pressure of CO2 in the atmosphere. ”
and the Revelle factor (and in turn the temperature of the oceans and its salinity), etc.
ATTP, I’m looking through your Python script plot_pCO2_DIC.py. I don’t have the python matlab library installed, so I can’t run it as it is right this moment. Please clarify meaning of variables: what are TA and CA? Why did you work in multiples of 1.0e-6 for DIC, CA, and TA? Am I correct in thinking that the entire inventory of carbon represented in your model is the atmosphere and the top layer of the ocean only? If not, where is the calculation for diffusion out of the top layer and into the deep ocean? Yours, Kevan
ATTP, Scratch that last question: you are calculating the equilibrium pH, so no need for any dynamics. But would appreciate clarification of variables. I am running the code now, without graphics, just printing the output and plotting in spreadsheet. Works fine. I’m not sure how to check it for you, but I’ll try. Kevan
I suspect TA and CA are titration alkalinity and carbonate alkalinity respectively. See section 1.2 of Zeebe and Wolf-Gladrow (seriously, see if your university has a copy in its library and borrow it, you will save yourself a lot of time and effort).
Yes, TA is titrate alkanity and CA is the carbonate alkalinity. The numbers are from measurements. The pre-industrial DIC is around 2002 micro-mol/kg. The 10^{-6} is because it needs to be converted to mol/kg for the actual calculation. As my other post says, the TA is 2311 micro-equiv/kg (I did work out exactly what that meant, but have now forgotten).
Okay, I think micro-equiv/kg just means the equivalent to micro-mol/kg.
Dikran,
It isn’t only seawater in which H+ ions exist as H3O+ , its true of any aqueous acidic solution. As you rightly point out H2O is a polar molecular and that allows weak intermolecular charge-transfer bonds to form between ions and water. This makes water a very solvent for salts (species that dissolve into ionic components) – as an example – the blue colour created by Copper salts (e.g Copper Sulphate) is entirely due to the species Cu(H2O)6 ++, which actually precipitates out with the water molecules still attached – see here.
Water, given its unique hydrogen-bonding capabilities, exists as a kind of extended “multi-molecule” not dissimilar to the structure of diamond, except that the single strong covalent bond between carbon atoms (C-C) in diamond is replaced a O-H…O arrangement (where – is a covalent bond and … a hydrogen bond). in water This, I think, allows point-like charged particles (ions) to be readily accommodated into this structure whereas hydrogen bonds from irregular covalent structures such as H2CO3 would disrupt this lattice.
“micro-equiv/kg just means the equivalent to micro-mol/kg.”
In this context, an equivalent means the amount that can react with one mole of H⁺. So that is one mole of HCO₃⁻ but a half mole of CO₃⁻⁻, since they are titrating to pH 4.5.
Nick,
Ahh, thanks. That explains why the TA is given by
Thanks Phil, good to know my inexpert intuition wasn’t too far off! ;o)
I’m aiming to read the Zeebe and Wolf-Gladrow book cover-to-cover, in the hope that the first reading will at least show me the key things I don’t know I don’t know, and hopefully to actually understand the material on the second.
A few points from General Chemistry 2.
When talking about equilibria use minimum free energy, not energy as the driver. The former includes entropic effects.
The reason for using [H+] is a) it is simpler and b) you don’t have to keep carrying extra water as in
HA (aq) = H+(aq) + A- (aq)
rather than
HA (aq) + H2O (l) = H30+(aq) + A-(aq)
Everybunny knows that there are no naked protons in solution, but H3O+ is also a simplification of the hydrogen bonding holding constantly forming and falling apart complexes of water and protons together. Of course the second one is needed when talking Bronsted Lowry conjugate acids and bases. It is also a good idea to label all the phases when dealing with solution equilibria because pure liquids and solids don’t participate in the equilibrium expressions as their concentrations remain constant.
Strictly speaking one should use activities not concentrations but for dilute solutions with oom answers this is usually not necessary.
Apologies, but the blackboards were screaming
“Everybunny knows that there are no naked protons in solution” thanks Eli, bunny’s may all know that, but apparently not all wombats!
Or BBDs…
TA and DIC correspond to the things that you look at when balancing equations. They are conserved. DIC is of course total C, and TA is (net negative) charge, limited for convenience to things that actually react here and appear in equations (so don’t count Na⁺ etc). So if you add H⁺, then TA decreases correspondingly, but DIC is unchanged. And if you add (uncharged) CO₂, then DIC increases, but TA is unchanged.
The interesting answer to what aqueous concentration Henry’s Law refers to, is at bottom a practical one related to how it is measured. Simply speaking a degassed volume of liquid is placed in contact with a known volume of gas at a measured pressure and the bunnies go out for a carrot, returning when an equilibrium has been reached and finally measures the equilibrium pressure. Thus, the Henry’s Law constant refers to the total number of moles of CO2 that have dissolved per liter of water and does not account for the chemical equilibria.
Important also to remember that Henry’s law assumes an equilibrium between a closed volume of gas in contact with a liquid, but that the oceans are not closed and as Nick said above, an equilibrium of that sort is never established.
This post has caused a reshuffle in my understanding of the meaning of acids and pH .
I sort of assumed water was at a pH of 7.0 and if the chemicals added to it were acidic or basic then the solution became acidic or basic .
Now I find that the pH we talk about for water is much more related to the atmosphere it is in and the pressure and temperature prescribed and that water as we know it does not have a true pH.
As Eli says above.
Hence rainwater is intrinsically acidic.
CO2 in air into raindrop makes it acid.
Nick might be able to explain what uncharged CO2 means as it appears that CO2 does have a charge when it goes into water.
Natural water at a natural pressure in natural air at 25C is not pure water though when specified this way it has a pH of 7.0.
This is because of the CO2 already in the air and water.
It is really the pH of a carbonated solution in the first place.
ATTP: I do not understand what you mean when you say, “The red line is the above calculation, while the blue line is the Revelle factor determined from the data plotted in the upper panel.” Where did the data for the top panel come from? If it came from your program, why are the two Revelle factors in disagreement?
I’d like to turn off the CO2 climate sensitivity effect in your code and get the graph of pC02 vs DIC from ionic balance alone. I am looking for the constant 3.0 C/ppmv you mention in your text, but don’t see it. Instead I see:
dF = 5.35*math.log(pCO2[i]/280.0)
dT = dF*0.75
Temp = (14.5 + 273.15) + dT
May I assume that setting dT to zero above will remove the temperature effect and otherwise leave the code undisturbed?
I would like to plot pCO2 vs DIC under the assumption that there is no acidification effect. I think that will take some more work, but I’ll try to do it myself by editing your code. The plot would allow us to see more clearly the significance of the Revelle effect in the diffusion of CO2 from the atmosphere to the deep ocean.
Raindrops are mostly acidic because of SOx pickup (countered a bit with amines picked up). CO2 acidification occurs when puddles stand for a long time People have actually studies this
http://www.sciencedirect.com/science/article/pii/S1352231008005980
Kevan,
All I mean about the red and blue lines is that I can calculate it from the equation that I give in the post, or I can approximate it using the pCO2 and DIC values that I’ve calculated.
As far as the ECS is concerned, yes and an ECS of 3oC is a climate sensitivity of 0.75 K/W/m^2.
I don’t think what you suggest at the end of your comment makes sense. I don’t know how you can turn off the acidification.
angech,
Why is it that so many who question our current understanding of this topic haven’t bothered to familiarise themselves with even the basics?
FWIW I think “Natural water at a natural pressure in natural air at 25C is not pure water though when specified this way it has a pH of 7.0. is wrong, pure water (i.e. nothing but H20, say in a sealed unreactive container with perhaps a noble gas atmosphere above) would have a pH of 7 IIUC. Natural water is mildly acidic, i.e. pH < 7.
“Nick might be able to explain what uncharged CO2 means as it appears that CO2 does have a charge when it goes into water.”
CO2 doesn’t have a charge when it is in water CO2(aq), but it reacts with H2O and dissociates into ions which are charged, e.g. H+ and HCO3-, but note if you add a + and a -, the result is uncharged on average.
Not the best of bases for the contention
… that the pH of the ocean exists in a reasonably narrow band and that concerns re rapid and dangerous pH changes in response to increasing CO2 rely on more than a simple surface exchange mechanism..
sed s/is wrong/is wrong (or at least very badly worded)/g ;o)
angech
Please stop the constant whine of contrarianism. We know from palaeocean evidence (sediment cores) that acidification occurs in response to pCO2 increase. Perhaps you could use these threads as an opportunity to learn more about the mechanisms rather than an opportunity to intrude your pseudoscepticism into the discussion?
ATTP,
“I don’t think what you suggest at the end of your comment makes sense.”
I want to see how 8 kg of carbon-14 that are created by cosmic rays get from the upper atmosphere into the deep ocean every year through a top ocean layer that has relative carbon-14 concentration of 0.96. I started with a linear model, in which the rate of emission from the ocean into the atmosphere is proportional to the amount of carbon in the atmosphere. The acidification of the ocean introduces a non-linearity into the carbon cycle, because the rate of emission is not exactly proportional to DIC as pCO2 rises. I want a clear and graphical representation of how large the effect of this non-linearity will be upon the rate of emission of carbon from the ocean as we increase the amount of carbon in the atmosphere. It seems to me that a comparison of pCO2 vs DIC with and without the effect of acidification will show us the significance of the non-linearity.
Kevan,
Have considered the biosphere in your model of carbon-14 uptake? At the moment, the biosphere is taking up about 25% of our emissions, so presumably must be taking up some of the carbon-14 too.
Kevan “I want to see how 8 kg of carbon-14 that are created by cosmic rays get from the upper atmosphere into the deep ocean every year through a top ocean layer that has relative carbon-14 concentration of 0.96.”
It appears that you still don’t appreciate the distinction between residence time and adjustment time. The 14CO2 isn’t removed from the atmosphere, it is exchanged with 12CO2 and 13CO2 from the other reservoirs, which doesn’t require a significant change in ocean chemistry. The removal of an excess of CO2 (e.g. from fossil fuel emissions) on the other hand means that the amount of carbon in the oceans must increase, and then the chemistry & Revelle effect becomes important. I seem to recall explaining this to you repeatedly on your blog and it seems that you have still not grasped the fact that the decay of bomb carbon and the decay of anthropogenic emissions are not governed by exactly the same processes.
Yes, but the biosphere’s carbon-14 concentration is 99% to 100% of the concentration in the atmosphere, so it cannot transport carbon-14 annually out of the atmosphere. I could add a little side-reservoir for the biosphere, but I don’t know what observations I’d test its behavior against.
Do you agree that it would be interesting to see pCO2 vs DIC for the simple linear model and the more realistic non-linear Revelle Model?
ATTP, I suspect that most of the 14CO2 taken up by land based plants is from seasonal primary production/decay, in which case the 14CO2 gets released back within a few years, which I suspect is one reason the decay of bomb carbon is rather slower than the basic residence time. It does need to be included in a model to get the answer right for the right reason (c.f. Enting and Pearman model – note the E&P paper explicitly points out that you need to model the raw fluxes between atmosphere and ocean/biosphere separately, rather than just the net flux, in order to get the 14C response and the adjustment time right simultaneously).
Kevan,
Possibly, I’m just not sure what it would mean if the simple model doesn’t include many of the processes that likely take place.
“Do you agree that it would be interesting to see pCO2 vs DIC for the simple linear model and the more realistic non-linear Revelle Model?”
I have to say I can’t see the point in this, other than to demonstrate the difference between a hopelessly naive “spherical cow” carbon cycle model and a model that is a bit more realistic (which can at least show why bomb carbon and pulse of additional CO2 would not decay at the same rates).
Dear ATTP, Another graph I’d like to see is the pCO2 vs DIC for no source of carbon other than CO2, so we can see how the CaCO3 changes the slope of the curve. I just received some of those four-pad pH strips. My tap water is pH = 8. Some bubbly water is pH = 5. According to a graph in a text book I’m looking at, the equilibrium partial pressure of CO2 for this water is 0.01 atm. I let the bubbly water sit for a while until it loses most of its initial fizz. I put some vinegar in. Now pH = 4. I think it’s more bubbly. I’m not certain, but when I move the strip around in the water, it’s looking like new soda. This suggest that the higher pH increased the equilibrium partial pressure for my soda water, thus encouraging more CO2 to come out. Based upon these crude observations, it seems to me that there will be a non-linearity in the CO2 exchange even without any dissolved CaCO3. I’m wondering if the dissolved CaCO3 has a significant effect upon the non-linearity or not. So, I will either figure out how to modify your code, or start from scratch myself. Obviously, my ideal outcome is you saying, “How interesting Kevan, yes, I think I’ll edit my own code myself.” But I’m not going to look a gift horse it the mouth. Thanks for your great piece of code. Yours, Kevan
BTW Essenhigh, and Starr and a host of others have already published papers on the spherical cow approach, to the extent that the IPCC SAR has a subsection to explain why such arguments are known to be wrong (as I mentioned on Kevan’s blog).
Kevan,
Firstly, the only source is CO2. The other inorganic carbon compounds came about because of the reaction of CO2 with water. Secondly, there is no CaCO3 in this model. That is another set of reactions that could be included. Nick Stokes has a post that includes this reaction. Eli also has a post that mentions it. However, as the quote at the end of my post indicates, the drawdown time due to the interaction with CaCO3 is thousands of years.
“Eli Rabett says November 5, 2016 at 2:53 pm
Raindrops are mostly acidic because of SOx pickup (countered a bit with amines picked up). CO2 acidification occurs when puddles stand for a long time People have actually studied this”
Yet the first reference I find is,
“Why is rain water naturally acidic?
Rain water absorbs carbon dioxide from the air it passes through on its way down. The carbon dioxide reacts with the water to form carbonic acid. This mechanism is one of the principal ways in which atmospheric carbon is scrubbed from the air and put back into circulation among living systems. Carbonic acid is only weakly acidic.The natural acidity of rain is due to the trace amounts of carbonic acid present in aqueous solution. It is not to be confused with the kind of acid rain that is sometimes the result of excess sulfur dioxide and other industrial pollutants in the atmosphere. Sulfur dioxide reacts with rain to form sulfuric acid”
Eli, your article is related to air pollutants/particles, not the natural pH of rain water in general which is relatively acidic due to CO2.
What do you think the pH of rain water range, unpolluted is?
Dikran Marsupial says:November 5, 2016 at 3:54 pm
FWIW I think “Natural water at a natural pressure in natural air at 25C is not pure water though when specified this way it has a pH of 7.0. is wrong, pure water (i.e. nothing but H20, say in a sealed unreactive container with perhaps a noble gas atmosphere above) would have a pH of 7 IIUC. Natural water is mildly acidic, i.e. pH < 7.
Nick might help,
PH of water would be acidic using the rainwater analogy above but I thought the definition had to do with water at a certain temp and pressure in air hence would have to have CO2 already in it.
Your definition makes much more sense in that it removes any component other than water.
So a mole of water has half a mole of H+ and half a mole of OH- or a mole of each and is neutral?
Adding CO2 reduces the acidity towards 4.5 and a mole of CO2 to a mole of water could theoretically make a mole of Carbonic acid.
Can this be described as a pure acid and is it the same as aqueous CO2 in this situation.
The pH of most of the water in the world is basic at 8.1 a high level away from the 5.5 assumed for a CO2 of 400. Hence other causes than the level of CO2 in the atmosphere are also at play in the acidity of ocean water overall and at the surface level.
Can I state that increasing CO2 definitely causes increased acidity of the surface level of the ocean. I have said this all through this and the preceding post. Anyone disputing this please put up a reference of mine or desist.
Thanks.
Angech: Rain water is not rain drops. Nucleation and growth of rain drops requires SOx (mostly as H2SO4.
Kevin H: You are confusing decay of C14 with dilution of C14.
Oh yeah, a mole of water has 1E-7 moles of OH- per liter and same of H3O+. Angech seriously go take GChem 2. Sea water is a buffered system, but its capacity to absorb CO2 and not change the pH is not infinite.
Dear ATTP,
“Firstly, the only source is CO2. The other inorganic carbon compounds came about because of the reaction of CO2 with water. Secondly, there is no CaCO3 in this model.”
I see. I was under the impression that a mixture of only CO2 and water will always be acidic. If we have CO2 and N2 over water, with CO2 at 400 ppmv, the pH of the water will be around 6, according to Figure 5.4 at link below, a figure I have seen in several places, and which is based upon the same ionic equations you have implemented in your python script.
Click to access Section5.pdf
I do not dispute the ionic balance you have calculated, but in order for the pH of the mixture to be alkaline (pH = 8.1 or something in this case), there must be something else significant going on in the water. One possibility is that it the ocean is saturated with CaCO3. According to Figure 5.6 at the above reference, at pCO2 400 ppmv, (Log(pCO2) = -3.4) over a solution saturated with CaCO3, the pH will be around 8 and the Ca++ concentration will be 70% of the HCO3- concentration. If so, then 70% of the DIC in the ocean arises from CaCO3.
Do you have another explanation for the alkalinity of the ocean?
Yours, Kevan
@-kevin
“Do you have another explanation for the alkalinity of the ocean?”
I don’t think anybody has another explanation for the alkalinity of the oceans because everybody who has studied this at least to chem2 level knows the observed data is accurate and has a comprehensive and detailed chemical explanation that is predicatively accurate and consilent with everything else we know and use in aqueous chemistry. Pick up on Eli’s hint and read up on Bronsted Lowry conjugate acids and bases, it might correct some of the assumptions that seem to be behind your confusions.
All this inorganic stuff just forms the substrate in which the biological activity takes place. precipitation of calcite skeletons by everything from sponges to coral is an enzyme mediated process that is significantly faster at removing CO2 from solution (in the various meta-molecular ion-water forms it takes).
The assumption seems to be that it would be possible to remove the cumulative carbon we have added to the atmosphere by removing atmospheric CO2 equal in amount to the emissions. But this requires that biological sink to work in the new base chemistry at least as effectively as it does now.
Observational studies do not confirm this.
http://www.nature.com/nature/journal/v407/n6802/full/407364a0.html
“The present rise in atmospheric CO2 levels3 causes significant changes in surface ocean pH and carbonate chemistry4. Such changes have been shown to slow down calcification in corals and coralline macroalgae5,6, but the majority of marine calcification occurs in planktonic organisms. Here we report reduced calcite production at increased CO2 concentrations in monospecific cultures of two dominant marine calcifying phytoplankton species, the coccolithophorids Emiliania huxleyi and Gephyrocapsa oceanica . “
Kevan,
I don’t see how you get that from that figure. As I understand it, if you fix atmospheric CO2 at 400ppm, the pH of the ocean would be about 8.
My understanding of the reaction with CaCO3 is that if there is an increase in CO2 then that reduces the amount of CO3-. The CaCO3 reation is
As this post by Nick Stokes explains, if you add CO2, then you change the CaCO3 equilibrium with CO3–, which ultimately leads to the dissolving of CaCO3.
Just to be clear angech wrote “Can I state that increasing CO2 definitely causes increased acidity of the surface level of the ocean. I have said this all through this and the preceding post.”
however he also wrote
“My contention is that the pH of the ocean exists in a reasonably narrow band and that concerns re rapid and dangerous pH changes in response to increasing CO2 rely on more than a simple surface exchange mechanism.”
the “dangerous” (in the sense of causing ecological damage) pH changes are those that are ocurring in the surface waters and angech was clearly contending on the other thread that they are not happening due to increased CO2.
Angech clearly has not been saying this “through this and the preceding post”, but is just trying to keep the discussion going (i.e. trolling), and this sort of disingenuous behaviour doesn’t deserve a reply.
Let me make a point about the Revelle factor and the surface layer. If we stopped emitting now (after emitting about 600GtC) we would expect the atmospheric CO2 to decay to a long-term value of about 330ppm. Based on my calculations above, the DIC would increase from about 2000micro-mol/kg, to about 2030 micro-mol/kg (an increase of about 1.5%) and the pH would drop to about 8.1. The oceans would have taken up about 75% of our emissions – so about 450 GtC. If this is 1.5% of the total amount of inorganic carbon in the layer that exchanges with the atmosphere, then the total would be 450/0.015 = 30000 GtC. This is almost as much as in the entire ocean, so it’s not simply a thin surface layer that is involved.
Eli Rabett says:November 6, 2016 at 2:09 am
” Rain water is not rain drops. Nucleation and growth of rain drops requires SOx (H2SO4.”
Eli,
Minute amounts for seeding. Rain drops are rain water.
I keep finding references that the pH of rain water and rain drops is usually in the vicinity of 5.5 and the cause is the CO2 going into the raindrop.
“The technical definition of pH is that it is a measure of the activity of the hydrogen ion (H+). It is essentially a measure of acidity. Normal rainwater has a pH of 5.6 (slightly acidic). This is because it is exposed to the carbon dioxide in the atmosphere. The carbon dioxide gets dissolved in the rainwater and forms carbonic acid (H{-2}CO{-3}).
Rainwater with ph value below 5.6 is considered as acid rain. There are both natural and non-natural sources of materials that cause pH of rain water to change. The primary air pollutants are sulphur dioxide and nitrogen oxide.”
If one thinks about it the CO2 going into the raindrop is doing the same thing as the CO2 going into the surface layer of the ocean.
Same CO2 in the atmosphere, same dynamics.
You cannot deny the role of CO2 in the raindrop and that ATTPs calculations would give it a pH of 5.5 under normal atmospheric conditions.
Why is there a problem with you accepting this?
Why do easily accessed references all say carbonic acid is the cause ?
I don’t know much about rain, but the first figure in this plot might be relevant. The pH depends largely on the DIC, so maybe a small raindrop can have a larger DIC value than the ocean.
“I’m not certain, but when I move the strip around in the water, it’s looking like new soda. This suggest that the higher pH increased the equilibrium partial pressure for my soda water, thus encouraging more CO2 to come out”.
What they are describing is commonly referred to as a water bottle bomb. The bicarbonate anions react with protons from acid to produce CO32, which decomposes to form CO2, and the dissociated CO2 is then released from the solution as the equilibrium between CO2(aq) and CO2(gas) changes. This can generate enough pressure to explode a plastic bottle. The experiment does not show that Henry’s constant depends on pH. Henry’s constant was the same before and after.
“Thus, the Henry’s Law constant refers to the total number of moles of CO2 that have dissolved per liter of water and does not account for the chemical equilibria”
Henry’s constant applies to all dissolved species of CO2. As the Handbook of Chemistry says: “Solubilities for gases which react with water, namely ozone, nitrogen, oxides, chlorine and its oxides, carbon dioxide, hydrogen sulfide, hydrogen selenide and sulfur dioxide, are recorded as bulk solubilities; i.e. all chemical species of the gas and its reaction products with water are included”. If Henry’s constant for CO2 were dependent on the relative concentrations of DIC (which changes with concentration) then kH would not be a constant for a given temperature. The constant does not change with concentration. It is a linear law. Kevan is correct.
Dear ATTP,
“As I understand it, if you fix atmospheric CO2 at 400ppm, the pH of the ocean would be about 8.”
That’s because the ocean is a saturated solution of CaCO3. If it were only CO2 and water, it would have pH = 6. I have some soda water going flat here, pH started at 4 and its getting close to 6 now (but I’m accurate only to +-0.5 with these pH strips). You can use the log(H+) concentration in Figure 5.4 below, derived from the same ionic equations you presented.
Click to access Section5.pdf
There is no way for the CO2-water system to generate a surplus of OH- over H+. I ran your program with initial DIC = 1000 rather than 1800. The equations tell us pCO2 is 0 when DIC = 1000. At DIC < 800 the program crashes with a divide by zero. Any model of CO2 mixed with water should give you pCO2 = 0 with DIC = 0. When you apply pH = 8 to those equations, you must be violating one of the ionic assumptions upon which the equations are based, because those same equations are used to deduce the H+ concentration, and hence the pH.
When you add Ca++ and CO3–, assume saturation, and add this into your ionic balance, then you can account for the ph = 8 of the ocean. From there, you can look at how the carbon concentration of the ocean increases with pCO2. Figure 5.6 at the link above does this. You will see that, for pCO2 from 1 ppm to 1,000,000 ppm, the DIC increases in proportion to pCO2, except that 70% of the increase is coming from an increase in Ca++ and the other 30% is coming from CO2 dissolving (I'm getting the 70% from the small offset between the HCO3+ and Ca++ graphs). Given that 70% of the DIC is due to Ca++ we see that the amount dissolved in the ocean that comes from CO2 increases in proportion to pCO2, making a linear diffusion model a good fit for ocean absorption.
Yours, Kevan
Yes, because Henry’s Law constant relates the amount of Dissolved CO2 and the partial pressure in the atmosphere above. In order to determine the actual concentrations, you also need to consider all the other reactions, which have their own constants. That’s all explained in this post. Essentially, these are a set of coupled equations that you need to solve together in order to determine the equilibrium concentrations.
If you wanted Henry’s Law to apply to all the dissolved species of CO2 it would then depend on the relative concentration of DIC. That it doesn’t is why it doesn’t depend on DIC.
No, he’s not and neither are you. Asserting otherwise doesn’t make it true.
Dear ATTP,
The ionic balance dictates H+ and pCO2 from DIC. For each pH, there is a unique value of DIC and pCO2. You cannot force H+ to one value, and then input a value for DIC and expect to get pCO2. When you operate the equations without over-contraint, they all work out to obey the classic Henry’s Law of proportionality of dissolved concentration with pressure. It has to be that way because, as I argued above, all forms of dissolved carbonate are in equilibrium with one another, so they communicate freely with the atmosphere through the CO2 (aq) form.
Yours, Kevan
Yes, I know; I haven’t done any such thing and it is essentially the point. The DIC is largely set by how much inorganic carbon is in the ocean. To get a pH of 6 would require a DIC of about 3900 micro-mol/kg, which is a bit less than twice what it is currently measured to be and would suggest that there would need to be twice as much inorganic carbon in the ocean than there currently is.
CO32 in my post should have read H2CO3
Dear ATTP, I don’t follow you. The pH of a CO2-water system with pCO2 = 400 ppmv is 6. Your equations should show this to be the case, although I have not tried to re-arrange your code to get pH from pCO2, and allow DIC to be calculated also. Regardless of pCO2, the pH can never be greater than 7. Assuming pH = 8 is, under all circumstances, a violation of the equations, and when so violated, the equations will give you nonsense answers. One such nonsense answer is, “The annual probability of a dissolved carbon atom being returned to the atmosphere decreases as the number of dissolved carbon atoms increases.” Yours, Kevan
Kevan,
I’m not following you at all. My equations do not seem to show what you claim that they show. Where are you getting your claim that the pH should be 6 if pCO2 = 400ppm?
This would appear to be very obviously not true.
“If Henry’s constant for CO2 were dependent on the relative concentrations of DIC (which changes with concentration) then kH would not be a constant for a given temperature. The constant does not change with concentration. It is a linear law. Kevan is correct.”
No he isn’t. You can see this if you consider a solution of NaOH, which is used for scrubbing CO₂ from air. It brings no DIC, but CO₂ is vastly more soluble in that than it is in water. But only as long as it lasts. By the time the solution has been converted to NaHCO₃, the solubility (of further CO₂) returns to the more moderate value in water. If you did Eli’s standard flask-shaking experiment with a little NaOH in the water, you would find that the apparent Henry’s Law constant was not constant at all. That’s because you would be confusing DIC with dissolved CO₂. Even a solution of Na₂CO₃ has much more affinity for CO₂ than does water, despite the fact that it starts with high DIC.
Dear ATTP,
I have not checked your equations, other than to look at the output when I change the input. Your initial assumptions appear to be the same as in the texts I have looked at. The fourth-order equation that results from CO2 and water only has been solved and plotted in Figure 5.4 below.
Click to access Section5.pdf
Als, from same text, “Therefore, in the CO2- water system there is never a net excess of base.” Furthermore, I have performed my own experiments: the pH of CO2 dissolved in water ranges from 4 to 6 as soda water goes flat. If your equations are telling you that the pH of CO2 dissolved in water is >7, your equations are wrong.
Yours, Kevan
Kevan,
Okay, I think I see the issue. As I understand it, that figure is produced on the basis of the Titrate Alkalinity (TA) being 0. So, if I understand it correctly, in the absence of another source then you would be correct. The existence of CaCO3 means that it is also in equilibrium with CO3^{2-} which means that the TA in our oceans is non-zero. So, yes, you’re right that you do need CaCO3 to procuce a non-zero TA, but that is expected to be roughly constant on relevant timescales and so you don’t really need to include the CaCO3 reaction in order to determine the equilibrium carbonate concentrations. So, all of my calculations are on the basis of TA being 2311 micro-equil/kg.
Richard, Nick, Richard is right when we consider a sample of pure water, but under those circumstances, given enough CO2 in the air, IIRC the pH will be so low that most CO2 in the water is in the form of CO2 anyway. Sea water isn’t pure, and as Nick alludes to, the pH set by other reactions is of major importance to the DIC. As correctly concluded by Kevan earlier on this thread, albeit a bit reworded, Henry’s constant would also have to include a pH dependence, if you want to claim it applies to DIC under all circumstances. In that case it is generally referred to as the effective Henry’s constant. The latter will fit quite nicely with the ‘true’ Henry’s constant when the pH is below 4.5, but above that value it rapidly changes.
ATTP,
If I understand correctly, in your simulation, you took the CO2-water balance and added OH- (NaOH for example) until the pH was 8.1. Then you calculated what the total dissolved inorganic carbon would be for this system as a function of pH. If so, there is still a problem with the calculation, because for pCO2 = 0, your calculation gives DIC = 1000 mmol/ml.
There are dozens of papers from the 20th century presenting measurements of CaCO3 saturation concentration in the ocean, as part of efforts to understand how CaCO3 precipitates as ocean water changes in depth and temperature. The ocean is not always saturated with CaCO3, but it is always close to saturation. So it seems to me that the simplest explanation for pH = 8 in the ocean is the obvious one: dissolved CaCO3.
If you add Ca++ and CO3–, the presence of the same CO3– group being contributed by both Ca++ and dissolved CO2 requires that you include the CaCO3 concentration balance in your ionic equations. This would be much easier for you to do than for me, because you have already programmed the CO2-water equations. But we could trust the solution presented in Figure 5.6 of the page I keep quoting.
I am now going to try your code with TA = 0 and see if it matches the plots in the Figure 5.4.
Yours, Kevan
Kevan,
Not really. I solved the equations based on the measured values for DIC and TA (you could try reading my earlier post where I explained this). I suggest that you read Zeebe (2012) and look at Figure 1; CO2 invasion/release can change the DIC, but not the TA.
I suspect that that is because you can’t really have pCO2 = 0 with TA not equal to 0. As I said above, the equations are being closed by specifying TA.
ATTP: With TA = 0 your code gets stuck in the loop where you calculate the change in H+ concentration for each value of DIC. Apart from a couple of bugs, this is a neat piece of code: it does a lot with very few lines. I might have to download that Matlab extension. My institute owns a site license. Yours, Kevan
Kevan,
Yes, that’s – I think – because the assumption here is that you’re in a regime where you can ignore [OH-] and so you can estimate CA from TA and the concentration of Boron.
“I suspect that that is because you can’t really have pCO2 = 0 with TA not equal to 0.”
It could be that I don’t actually understand what TA is. I thought it was the capacity of a solution to neutralize acid. I seem to remember placing a litmus strip in a solution whose TA we wanted to measure, then titrating acid into the solution until the litmus paper showed acidity. This capacity to is always zero for the CO2-water system in the absence of CaCO3.
Kevan,
Possibly, but we’re not considering a situation where there is no CaCO3. Also, if you read Zeebe (2012), it seems that the value for TA is not influenced by CO2 invasion/release, therefore you don’t need to explicitly consider the CaCO3 reaction if you just want to determine the pH and concentrations of the various carbon species.
“it seems that the value for TA is not influenced by CO2 invasion/release”
If TA is the H+ concentration we must add to neutralize the solution, adding CO2 will reduce TA because CO2 adds H+. From my reference, “The system can neutralize added base, but can not neutralize added acid.”
“you don’t need to explicitly consider the CaCO3 reaction if you just want to determine the pH and concentrations of the various carbon species.”
I’m going to think about that, but it does make sense. You only have to worry about the CaCO3 if you allow more CaCO3 to dissolve, because then you have the solubility product of CaCO3 to deal with.
I’m still not sure what you are simulating. Suppose we define the following:
[DSC] = dissolved solids carbon concentration
= carbon from dissolved solids like CaCO3
= carbon concentration for pCO2 = 0
[DGC] = dissolved gaseous carbon concentration
= carbon from CO2 gas
[DIC] = [DSC] + [DGC]
In your simulation, what is [DGC]?
In a system of only CO2 and water, which will always be acidic, [DGC] is proportional to pCO2 (Figure 5.4). In a system of saturated CaCO3 with water and CO2, [DGC] and [DSC] are both proportional to pCO2 (Figure 5.6).
You appear to be starting with a saturated solution of CaCO3 with pCO2 = 400 ppmv, which has pH = 8. Now you increase pCO2 but do not allow more CaCO3 to dissolve. You calculate [DIC]. I want to see [DGC]. You cannot decrease pCO2 because the CaCO3 will start to precipitate out. I suggest you disable boron, nitrogen, and temperature effects in order to isolate and debug the CO2-water ionic equations.
I claim that, no matter what you do, so long as your equations represent physically possible solutions, the [DGC] will always be proportional to pCO2.
Kevan,
It appears that you haven’t read Zeebe (2012)? Why don’t you start there. What I’m presenting here isn’t really my calculation, it’s pretty much the standard one. It doesn’t include the dissolution, or formation, of CaCO3 (which is why it assumes that the TA is fixed) but – as I understand it – that is a process that draws down atmospheric CO2 over kyr timescales and can therefore be ignored if you’re simply interested in the basic of CO2 dissolution in seawater, which is relevant for decade/century timescales.
“Now you increase pCO2 but do not allow more CaCO3 to dissolve”
Not sure what you are getting at, Kevan, but in a situation with saturated CaCO3, this is not possible. [Ca2+]*[CO32-] is constant in a saturated solution (the solubility product). In other words, CaCO3 can only dissolve if some of the carbonate already in solution is transformed into bicarbonate.
“It appears that you haven’t read Zeebe (2012)?”
I took a look at Zeebe (2012). I did not find it as informative as the reference I have been quoting: no graphs of the ionic concentrations with pH nor presentation of the fourth-order equations, and containing occasional unsubstantiated statements like “This rule is frequently ignored, which has led to misinformation in the literature.” Not my style.
“in a situation with saturated CaCO3, this is not possible. [Ca2+]*[CO32-] is constant in a saturated solution”
I guess I was assuming that CO2 can contribute CO3–, so if [Ca++]*[CO3–] is constant, [Ca++] must reduce. I was also looking at Figure 5.6 in my reference, in which Ca++ drops with falling pCO2. As we increase pCO3, the CO3 starts to convert into HCO3, which permits Ca++ to increase. Note that, according to the ionic equations for saturated CaCO3 solution, the peak of CO3– concentration occurs at pH = 6, while in the CO2-water only system, CO3– becomes dominant only for pH > 10. For these reasons, it seems to me that the CaCO3 does affect the ionic balance of the CO2-water system.
If you fix the CaCO3 concentration and increase pCO2 you will have different equations, for which I have no plots. So far as I can tell, that’s the system you were attempting to simulate. I claim that if your simulation runs properly, [DGC] will be proportional to pCO2. But I am ready and willing to be proved wrong.
Yours, Kevan
Oh: I see that my double-dashes, which were suppose to indicate 2-, come out as one long minus sign, which explains why you write “2-“. Will do the “2-” in future. Or I could try CO3- -.
https://en.support.wordpress.com/latex/
Kevan,
Except that was quite a key point. It was related to something I said in my earlier post. You essentially have 4 equations with 6 unknowns, and so you need to specify two other parameters. To understand the carbonate chemistry of the ocean, it is often DIC and TA, both of which are conserved.
Kevan,
I’m not entirely sure what you’re suggesting, but the bit of Zeebe (2012) I was hoping you would read is the section on timescales. On geological timescales, the atmospheric CO2 concentration is basically set by volcanic outgassing (i.e., it is at a level where the rate at which CO2 is sequestered by the slow carbon sinks matches the rate at which it is outgassed by volcanoes). Therefore, over sufficiently long timescales, CO2 will be drawn back down to pre-industrial levels. However, that is expected to take > 100kyr. What I’ve done here is consider what I think is the relevant chemistry for decade/century timescales and can be used to have some idea of how much of our emissions will be taken up by the oceans on those timescales.
Dikran Marsupial says: November 5, 2016 at 3:54 pm
” pure water (i.e. nothing but H20, say in a sealed unreactive container with perhaps a noble gas atmosphere above) would have a pH of 7 IIUC. Natural water is mildly acidic, i.e. pH < 7"
Mildly acidic meaning 5.5 or thereabouts agreed.
Reading over at Nick's site comments indicate that the ocean is supersaturated by CaCO3.
Hence the pH of 8.1.
Natural water, raindrops, rainwater, and pure water, no contaminants but containing aqueous CO2 as the air has 400 ppm has a pH of 5.5
Ocean water is saturated with material from the earth's crust, in particular lots of CaCO3, which is why it is at 8.1 overall.
Increasing CO2 in the air has been unable to decrease the surface acidity by more than 0.017 per year.
Because of the high TA and supersaturated CaCO3 increasing CO2, which causes a drop in surface pH, is unlikely to move overall ocean pH outside a narrow range because there is always more renewable base to mop up excess CO2.
Ocean pH will change with CO2 increase and has changed for long periods with CO2 outgassing in the past.
angech,
My understanding is that on timescales of decades/centuries, the CaCO3 influence is relatively minimal and you can get a good sense of what will happen by assuming a constant TA. The reaction with CaCO3 draws down atmospheric CO2 on thousand year timescales.
“Natural water, raindrops, rainwater, and pure water, no contaminants but containing aqueous CO2 as the air has 400 ppm has a pH of 5.5”
angech if water contains CO2 it isn’t pure water.
“Increasing CO2 in the air has been unable to decrease the surface acidity by more than 0.017 per year.”
Yes, which is 0.17 per decade, or 1.7 per century; pH is a logarithmic scale, so while it may seem like a small number, that doesn’t mean it actually is (you need to show that ecological systems are tolerant of that variation). You provide no evidence whatsoever that this will not be “dangerous” (in the sense of ecological damage) in the medium-long term in contradiction to the world’s experts on the topic.
I am not interested in your trolling, especially given your repeated examples of disingenuous behavior.
Kevan wrote “This rule is frequently ignored, which has led to misinformation in the literature.” Not my style. good job not everybody is put of by style ;o)
Dear ATTP,
We can discuss geological timescales another time. I came here to discuss your calculation of [DIC] versus pCO2 for the ocean. I commend your effort to perform these calculations yourself, and I am impressed with the efficiency of your code.
Nevertheless, your code is wrong. It gets stuck in an infinite loop for TA = 0, it says [DIC] = 1000 ul/mole for pCO2 = 0, and it crashes for [DIC] < 800 ul/mole. If you want to correct and re-write it, please declare your intention, and I will help you debug the new code.
In the meantime, we have two examples of calculated ionic concentrations from traditional chemistry text books: one for CO2-water, one for CO2-water with saturated CaCO3. Both systems show that [DGC] = "combined concentration of all species of dissolved CO2 including CO2 (aq) H2CO3, HCO3-, and CO3- -" increase in proportion to pCO2. This may seem miraculous, but it is certainly the case, despite all the confusing complexity of the ionic equations. Both systems are presented below.
Click to access Section5.pdf
As Richard pointed out, the Handbook of Physics and Chemistry states unambiguously that [DGC] is proportional to gaseous partial pressure. So far as I am concerned, there is no higher authority in Physics and Chemistry than the Handbook. It has been examined and corrected for 97 years. Text books take second place to the Handbook. Published papers third place. As I have made clear to you elsewhere, published papers in Climate Science don't have any credibility with me whatsoever. The onus is on you to calculate the ionic balance for a physically-plausible ocean system in which [DGC] is NOT proportional to pCO2.
If you do not intend to correct your code, please say so, and I'll sign off and plan to solve the equations myself. If I ever get around to doing that, I'll send you a link and an invitation to come and debug my work, which I hope you will accept.
Yours, Kevan
Kevan,
I’ve already explained that my code is based on an assumption that one can ignore [OH-]. That’s why it gets stuck when TA = 0. That doesn’t make it wrong, it just makes it only suitable for situations when one can ignore [OH-].
Dear ATTP,
I’m taking that answer as meaning, “No, I’m not correcting the code, because it does not need correcting.” Very good. Thank you for answering all my questions, and for sharing your code with me.
Yours, Kevan
Actually, I don’t remember you ever saying this before, but I don’t saying so reflects particularly well on you. YMMV, of course.
Kevan,
Maybe read Marco’s comment. If the pH is low enough then CO2(aq) dominates the DIC and you would then have that the DIC is directly proportional to pCO2.
You wouldn’t be appealing to authority, would you?
Kevan wrote “So far as I am concerned, there is no higher authority in Physics and Chemistry than the Handbook. It has been examined and corrected for 97 years. “
This is an argument from authority, about which Kevan earlier wrote
“No scientist uses argument by authority. Science is based upon observations of nature, not worship of the word of experts. And yet argument by authority is all you have. Any time you try to debate me on actual numbers and calculations, you lose.”
which now seems somewhat ironic, given that he is dismissing ATTP’s calculations ;o)
“As I have made clear to you elsewhere, published papers in Climate Science don’t have any credibility with me whatsoever. “
This is what is known as an ad-hominem (an attack based on the source of an argument, rather than its content), which is essentially the flip-side of the “argument from authority” coin, which shows that Kevan is being a bit inconsistent here.
Most computer programs are written to operate correctly for a given range of inputs, rather than attempting to deal with all possible combinations. This is especially true when trying to convey a basic idea using a simplified model of the full system that is never going to be realistic beyond certain bounds. Dismissing ATTPs’ program as “wrong” just because it doesn’t consider the full range of conditions that Kevan wants is clearly unreasonable behaviour, and I suspect just a means of avoiding admitting that Kevan’s understanding of the carbon cycle is faulty. This quote from Kevan’s blog gives an indication motivated reasoning in operation:
“Your field has been able to deceive the public by exploiting the reputation science has earned through the efforts of people like me. You are not the first pseudo-scientific field to do this, nor will you be the last. Fields like yours are like tumors that grow inside the wonderful creature that is the scientific community. I am an antibody from the scientific community, doing my part to try to shrink you until you are harmless. I am here because I know that your experts are wrong, and that your field is cancerous.”
Dikran,
I’d forgotten about Kevan’s comment expressing his views about climate science.
Kevan “As Richard pointed out, the Handbook of Physics and Chemistry states unambiguously that [DGC] is proportional to gaseous partial pressure. ”
Can I have a page reference for that please?
I reread Richards comment (and checked the quoted bit in the Handbook)
“Henry’s constant applies to all dissolved species of CO2. As the Handbook of Chemistry says: “Solubilities for gases which react with water, namely ozone, nitrogen, oxides, chlorine and its oxides, carbon dioxide, hydrogen sulfide, hydrogen selenide and sulfur dioxide, are recorded as bulk solubilities; i.e. all chemical species of the gas and its reaction products with water are included”. If Henry’s constant for CO2 were dependent on the relative concentrations of DIC (which changes with concentration) then kH would not be a constant for a given temperature. The constant does not change with concentration. “
As far as I can see (from that) the handbook does not state unambiguously that [DCG] is proportional to gaseous partial pressure, but it is instead Richard’s inference based on a table of solubilities, which is not the same thing. Please can you give an exact quote for this with a page reference?
“Both systems show that [DGC] = “combined concentration of all species of dissolved CO2 including CO2 (aq) H2CO3, HCO3-, and CO3- -” increase in proportion to pCO2.”
Kevan, it’s not a linear correlation, which Henry’s constant says there should be, and that’s because of the role of pH. There’s a good example shown in the link below of Henry’s constant being, very, very much dependent on solution pH, in particular above pH 5 or so.
http://www.ems.psu.edu/~brune/m532/m532_ch5_aqueous_phase.htm
As noted here in a study on compiling Henry’s constant
Click to access jpcrd427.pdf
“…the ionization is quite small and has been neglected in this study”.
It’s fine that you put the Handbook of Physics and Chemistry as such an important source, but the Handbook does not take into account the effect of pH, or it would have to list a lot of values for CO2. Fortunately, there is a relatively ‘simple’ method to calculate the effective Henry’s constant using the equilibrium constants for the two acid-base reactions and the pH (read: hydrogen concentration) as shown in the first link.
If you really want to have fun, see:
http://onlinelibrary.wiley.com/doi/10.1029/2004JD005220/full
Oh wait, I forgot, you don’t believe any publications in the field of climate science…
Dikran, the funny thing is that the Handbook refers to my “nist” publication (link in my previous comment) for the CO2 data, which explicitly notes that ionization is ignored because it is so small. And that’s because the pH is so low when you dissolve CO2 in pure water…
Well, obviously.
Depressing thread, really.
Because I have nothing better to do 🙂 I modified my code to do as Kevan asked. The only thing I wasn’t sure of was the temperature dependence for the H+, OH- reaction, so assumed there wasn’t one and simply used . I also ignored Boron and assumed that
. I also ignored Boron and assumed that 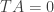 . The code is here, and the figure below is an inverted version of Figure 5.4 here.
. The code is here, and the figure below is an inverted version of Figure 5.4 here.
Hmmm, okay, the CO3^{2-} looks like its a bit too high. I’ll work out why later.
Looks like it might just be the temperature and salinity dependence of the coefficients.
Dear ATTP & Dikran,
While attempting to help Kevan improve his understanding of the Carbon Cycle is obviously a bit frustrating (if not infuriating at times) let me offer my thanks for for explaining things both here and at Kevan’s blog. I have found your explanations have helped my understanding greatly and am now pursuing several of the papers you have highlighted. Best Regards
David,
Thanks, glad someone found it useful 🙂
Cheers David, I’ve learned a fair bit from the discussion as well and ATTP’s two blog posts and code are a good resource.
ATTP
given that the pH is lower towards the pole and that CO2 dissolves better in colder water may explain this should there not be a polewards decrease in the CO2 levels in the atmosphere at the poles? Would you formulae concur with this or give the same CO2 in the air everywhere regardless of temperature.
angech,
I don’t know if it concurs with it, but it certainly depends on temperature (see the second figure in the post).
thanks
“given that the pH is lower towards the pole and that CO2 dissolves better in colder water may explain this should there not be a polewards decrease in the CO2 levels in the atmosphere at the poles?”
Not necessarily, in the south there is Antarctica, rather than open water and in both hemispheres there is an ice cap, which I would have thought would partially block the exchange between the oceans and atmosphere. Also the production of CO2 is not evenly distributed across the planet, for fossil fuel use, much of it is in the temperate regions of the Northern hemisphere (there certainly is a gradient in concentration between the hemispheres). Also as I understand it, ocean uptake also depends quite a lot on the depth of the mixed layer, which in turn depends on wind. Basically the point I am making is that we might confidently expect to see such a gradient on a “spherical cow” water-world model of the Earth, but that doesn’t mean we will definitely see one on the real Earth as there are undoubtedly many other factors involved. As Einstein (is often quoted as having) said, “Things should be made as simple as possible, … but no simpler”.
There is a network of monitoring stations, which includes sites at high latitude in both hemispheres, which should provide some evidence.
Dikran:
http://www.esrl.noaa.gov/gmd/ccgg/ggrn.php
There’s even a movie of how it has changed as a function of latitude over time, here:
http://www.esrl.noaa.gov/gmd/dv/iadv/graph.php?code=ALT&program=ccgg&type=lg
Correlation to local temperatures is rather limited…
Actual CO2 concentrations map:
Thanks Marco + VTG, making a realistic model of the carbon cycle is actually pretty difficult as there are a lot of components that individually seem to make sense, but considering only one at a time can be rather misleading.
The video in Marco’s second link is very good, it looks like the hemispheric gradient is dominant. It is interesting that levels are perhaps higher in the Arctic, it could be that the atmospheric circulation (Hadley cells, jetstream, polar vortex) etc also prevent a simple gradient towards the poles.
Pingback: No, stabilising emissions will not stabilise concentrations! | …and Then There's Physics
Pingback: 2016: A year in blogging | …and Then There's Physics
Pingback: Residual airborne fraction | …and Then There's Physics
Pingback: Oh no, not again | …and Then There's Physics
Pingback: A bit more about committed warming | …and Then There's Physics
Pingback: No, we’re not slipping into a proper ice age | …and Then There's Physics
Pingback: CO2 sequestration during glacial maxima | …and Then There's Physics
Pingback: The Carbon Cycle | …and Then There's Physics
Pingback: Cycle du carbone océanique et devenir de nos émissions de CO2 – CARBONE#2 – Le Réveilleur